
新药海外临床研究核查面面观
发布时间:2025-02-24 10:09:05作者:彭晓雯来源:医药经济报
中国创新药企业的国际化进程已迈入以研发和技术创新为核心的新阶段。不少企业已构建起包括自主研发、许可交易、合资和战略外包等多元化出海路径。在此过程中,药品的临床试验质量是不容忽视的重要一环。如何全方位把好质量关? 笔者所在企业为临床CRO(合同研究组织),2021年,我方曾和申办方药企一起,经历了美国FDA对新药临床研究质量的核查。当年3月,我方收到FDA核查通知,8月迎接FDA对申办方和CRO的质量体系的远程核查,9月和10月又相继迎接了两家研究中心的现场核查。
2021年3-10月,申办方、CRO和Site(研究中心)各方人员都付出了极大努力,最后顺利通过核查,没有发现重大的缺陷。笔者将分享令人印象深刻的几点感悟,以飧读者。
全面关注质量体系和TMF系统
在接触过程中,笔者感受到,FDA对申办方和CRO的质量体系和临床试验主文件(TMF)系统的核查认真且呈系统性。从2021年3月FDA通知药企要进行核查时,药企就接到通知,按FDA的要求提供和翻译电子TMF给FDA核查员陆续查看。当年8月中旬,2名FDA核查员和1名翻译通过电话对药企和CRO进行了为期一周的远程核查访谈。
在远程电话接触中可发现,核查员基于前期TMF文件提问很具体。例如:某监查访视报告里的某次发药错误后续如何解决;某中心某次方案违背相关的整改预防措施;某中心某个严重不良事件(SAE)通报如何改进等。
除了具体细节问题,核查员也询问了药企申办方和CRO许多试验重要的流程问题,例如临床研究监查员(CRA)如何选择和培训、研究中心如何选择、影像数据如何评估等。
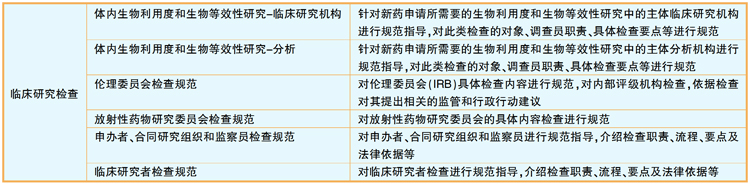
FDA对于临床试验数据的核查分为六小类:BA和BE研究(机构、实验室),伦理委员会,申办方+CRO+监查员,研究者(研究中心)等(详见上图)。在FDA官网首页的搜索栏输入“bioresearch monitoring, compliance program”,即可下载BIMO检查的各种指南。
如果药企计划未来药品出海到美国,笔者建议:
1.做好充分事前准备工作:了解FDA核查指南。
2.重视合作伙伴选择:如果将试验外包给CRO完成,需选择质量体系良好的品牌CRO,畅通FDA对CRO的核查。
3.重视TMF文件归档:在试验过程中,各方应通力协作,做好TMF归档。如果TMF文件产生后未能及时回收,可能会永久丢失。待到核查前再去补救,费力低效,最终咽下苦果的还是药企。
现场核查数据与流程情况并重
FDA在研究中心的现场核查,除了关注关键的试验入排数据和疗效数据,也非常关注试验流程及其遵循ICH-GCP和当地法规情况。
在某一研究中心,FDA核查员(仅一名,配一名翻译)面谈了主要研究者(PI)、次要研究者(sub-I)、临床研究协调员(CRC)、机构、伦理委员会,并仔细查阅受试者的病历,耗时10个工作日。
在研究中心,核查员尤其关注受试者每条入选排除标准和溯源的依据,每天都与sub-I沟通,发现的疑问主要包括以下方面。
GCP病历细节:部分病历未签字,该中心如何定义原始数据,如何录入数据?
外院就医记录:核查员尤其关注与入排有关的医疗操作,例如医嘱末页研究者和护士的签字情况。
患者外院放疗经历记录:研究者如何评估放疗量,是否违背了入排标准?
受试者外院影像上传完整性:有部分外院影像未上传给供应商,如何保证进行了完整的肿瘤评估?
核查员在与CRA访谈时,主要关注以下方面。
信息完整性和监查深度:CRA如何监查入排标准,查询了哪些资料?CRA在监查中是否发现信息存在不清晰、不一致的地方,是否采取了进一步行动与研究中心确认。
不良事件报告与处理:研究中心的可疑非预期严重不良反应如何递交?
监查工作的管理:CRA是否按规定频率与标准访视中心?CRO如何保存监查报告?
申办方稽查流程合规性:申办方的稽查流程、近几年如何完成中心访视、是否有沟通文件记录、是否有监查指导文件等。
需指出的是,核查员还尤其关注PI如何监管试验。
此外,核查员在另一研究中心询问了一些新问题,例如:
知情同意书(ICF)合规性:ICF中是否有可供查询临床试验登记信息的网址或平台;如果研究者发现了ICF中有缺陷,是否反馈给相关人员;中心有无反馈修正ICF的流程文件。核查员还请研究者复述知情同意的过程。
伦理资料完备程度:包括从试验准备阶段到递交伦理审核,和获得伦理委员会批准,全套的流程和各方如何与EC沟通的记录。
在这家研究中心,核查员还现场走访了药房、静配中心、样本储存室、离心室和机构物资室。核查员也仔细核对了受试者的逐条入排标准、仔细询问和查验了方案偏离的发现、报告和处理过程,对于影像评估的疗效数据,也是将受试者的影像学数据与上传到中心试验室的影像清单一一比对,根据疑点询问现场人员。
另外,核查员发现,该中心近几年受试者的随访、严重不良事件报告、方案偏离或药物超温等沟通邮件都未抄送研究者,仅仅发邮件给CRC。据此,核查员提问:研究者如何了解和及时处理这些情况。
注意核查结论性finding(问题)判定和落笔
FDA核查对于凡是已被解释和澄清过的问题,均不会在最终核查总结会或报告时再将其记录为一个问题。
根据现场参与的CRA同事提供的信息,研究中心核查,被落笔记录为FDA核查问题的只有3个:1)不良事件和合并用药漏录入EDC;2)计划外访视的影像未上传到中心影像;3)入排标准符合性未在病历上详细体现,记录不完整。
正如ICH E8(R1)指出的:申办方应前瞻性确定临床研究的关键质量因素并在试验过程中加强管理。这些关键质量因素包括但不限于:
知情同意是否恰当获得;
方案入排标准在招募时的执行情况,尤其是保证受试者权益的标准;
研究药物记录和管理流程;
与临床试验有效性终点相关或方案特定要求的安全性终点相关(例如严重不良事件、死亡、脱落等)的评估处理流程体系;
与临床试验的可靠性、完整性相关的流程体系(例如方案违背管理、盲态保持管理等)。
上述FDA核查显示:FDA高度重视关于受试者是否符合方案的入排标准和试验有效性终点数据和安全性终点数据,以及方案偏离的处理。
★★★ 小结 ★★★
综上,有计划将中国研发的新药出海到美国的药企,应前瞻性重视上述关键质量因素,选择质量体系可靠的CRO,选择可信赖的研究中心和研究者,扎扎实实遵循GCP规范。
不论是中国还是欧美的临床试验数据核查,都在考察试验过程的规范性、数据的科学可靠性,需要企业提前考虑和布局。
此内容为《医药经济报》融媒体平台原创。未经《医药经济报》授权,不得以任何方式加以使用, 包括转载、摘编、复制或建立镜像。如需获得授权请事前主动联系:020-37886610或020-37886753;yyjjb@21cn.com。





