
新药临床试验主动合规方能行稳致远
发布时间:2024-10-28 16:20:02作者:彭晓雯来源:医药经济报
远古时代,祖先们在自然界的植物动物矿物中寻找药物。“神农辨药尝百草”意味着找药者亲口尝试药物和检验药效。这种做法风险巨大:传说中神农尝百草多次中毒,最后因尝了断肠草而客死他乡。20世纪人类进入现代制药工业时代,如今一款新药上市,需要经过实验室、动物实验再到人体的Ⅰ期到Ⅳ期试验等逐步验证其疗效和安全性,上市后仍需按监管部门的要求,持续监测药物的不良反应。
一款新药的药效如何验证?现代的新药临床试验,从试验设计到执行,到最后迎接监管机构的核查,是如何保证药效数据确凿,避免它仅起到“安慰剂”作用?企业又如何主动合规,确保新药疗效大于安全性风险,提升上市成功率?
试验前
随机双盲,科学设计防偏倚
影响人体健康的因素繁杂,涵盖饮食、自身免疫力、心理因素等。因此,一款新药要想证明疗效确凿,必须过随机双盲对照试验这一关。
何为“随机”?即研究者随机让符合入选排除标准的患者,进到试验组和对照组,使他们在人口统计学(性别、年龄)和临床特征上具有可比性,从而减少潜在的混淆因素。
何为“双盲”?是指试验药物在外观上,与“安慰剂”和阳性对照药无差异;患者被随机分配到不同的用药组,受试者和医生、药师、护士都不知道某一位参与试验的患者吃的是“真试验药物”还是“阳性对照药”或是“安慰剂”,以避免心理暗示作用下,对药物真实效果有一定影响。
随机前,试验可能还会按实际情况进行“分层”。即研究者有时还会根据性别或年龄对受试患者进行分层,再随机,以确保没有性别、年龄或某些因素造成的疗效偏倚。
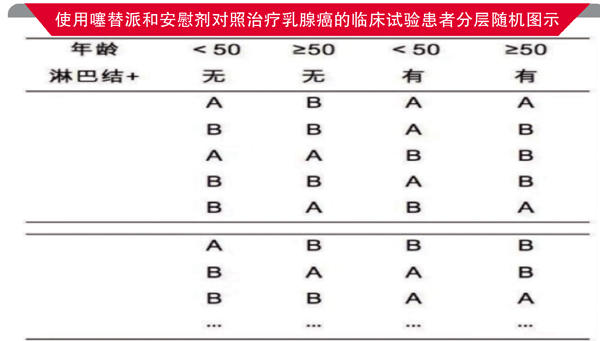
例如,下图显示,用噻替派治疗乳腺癌临床试验的患者分层随机安排,分层因素有两个:患者年龄是50岁以下还是50岁以上;乳腺癌病情是否有淋巴结的转移。因为年龄不仅会影响到术后生存时间,而且与免疫佐剂的疗效有关,50岁是否绝经情况也基本一致。
因此,随机分层后,这项研究的患者被分成四个类型:年龄<50,无阳性淋巴结;年龄≥50,无阳性淋巴结;年龄<50,有阳性淋巴结;年龄≥50,有阳性淋巴结,按1:1随机分的四组,使用试验药物A药和安慰剂B药的情况如图所示,通过这种方式分组服药后,再收集和对比几组患者的疗效数据,就可以最大程度减少偏倚。
试验中
严守GCP,监查稽查齐努力
新药开发在进行人体试验之前,应遵循药物非临床研究质量管理规范(GLP)。进入人体试验阶段,则应遵循药品临床试验质量管理规范(GCP)。与此同时,药企自身的CRA监查、QA独立稽查等也是保障新药疗效和安全性数据可靠的有力手段。
良好的GCP应包含以下要素:
1.试验方案明确详细,如有偏离,需要记录报告。
2.配备专业临床试验研究人员。申办方应建立临床试验质量管理体系,事先制定各种标准操作流程和试验计划书,并任命监查员到研究中心(医院)监查。
3.确保临床试验数据记录和报告准确、完整和可追溯,在研究中心和申办方保存临床试验的必备文件。对收集到的受试者相关数据,由各方进行仔细审核,确保数据准确性和一致性。
4.接受监查、稽查和药政核查。
5.做好不良事件报告工作。所有不良事件均应记录在源数据和电子化计算机系统中。安全性信息要进行定期汇总分析,制定药物安全风险管理计划;要对受试者用药后发生的不良事件进行及时的医疗救治或处理(减量停药或退出试验),确保参与者的安全。
6.妥善管理试验药物。药物的发放、接收、使用、回收、标签、温控都要有记录和明确的负责人和操作流程。
试验后
药政核查,聚焦关键数据
国家药监局2020年颁布的《药品注册核查要点与判定原则(药物临床试验)》(以下简称《判定原则》)对核查员如何判定临床试验合规性有详细的描述。简单来说,药监局核查员会核对以下问题的原始书面证据并访谈研究者、药师、监查员等。申办方应重点关注以下方面。
受试者入组合规性:参与临床试验的受试者是不是真实参与了试验?是否按试验方案的入选排除标准入组?入组用药前是否做了知情同意?
试验方案执行合规情况:受试者是否依照试验日程和剂量使用药物、进行化验或做安全性检查?是否按规定时间点做疗效评估(例如拍肿瘤的CT影像、抽血化验)?
不良事件和病历记录规范程度:核查员会核查是否病历和化验单上有(严重)不良事件漏报或未及时上报;试验室检查(如血生化检查值、肝肾功能指标等)的异常结果是否被适当评估和记录(如果是不良事件)?
生物样本和药物管理情况:生物样本(如血样)处理是否正确?药品接收、储存、随机发送、回收等是否符合规范,是否记录完整?
盲法试验规范管理:如涉及盲法试验,是否按照试验方案的要求设盲、保持盲态和实施揭盲;意外破盲或因SAE(严重不良事件)等需紧急揭盲时,研究者是否按照紧急揭盲规程操作并书面说明原因。
此外,核查员还会请申办方或CRO的医学监查员、数据管理、统计师到现场或远程协助回答试验的问题等。
现举一个糖尿病药物试验的例子说明,核查员如何保证受试者合格。
如果方案的入选标准显示:受试者筛选时,要符合WHO颁布的2型糖尿病标准,并且至少已连续12 周稳定使用二甲双胍片≥1500mg/天进行治疗。
那么,核查员就希望在所有受试者文件夹里,能看到既往外院病历(医嘱、处方)或该医院的HIS信息系统(医嘱、处方、付费记录)有该受试者被诊断为2型糖尿病的字样,并且还有购买二甲双胍片药的记录(发票、付款记录)。
如果方案的排除标准里写“应排除受试者有:不稳定的或快速进展性肾脏病史、筛选时处于活动性肝脏疾病期或明确诊断的精神疾病”。
那么,核查员就不能在入选的受试者病历或HIS里看到上述应该被排除的疾病诊断。
如果核查员看不到应有的2型糖尿病诊断证据,或发现理应排除的疾病出现在入选受试者文件夹。那么,核查员会认为该试验入选了“不合格”患者,相关数据应从试验最后汇总分析报告剔除。剔除后,如果达不到统计学意义上的受试者人数,或存在其他重大不合规情形(比如多次或多人使用试验禁忌药、或盲态未保持,甚至数据造假等),则会判为新药注册核查不通过,不批准该药的注册上市。
我国临床试验的数据质量正日益提升,近几年,国家药监局的年度核查报告显示,绝大部分临床试验数据可靠,合规良好,可顺利通过核查。
需强调的是,制药企业作为新药研发的主体,应始终坚持以患者福祉为先的伦理和科学原则,切勿抱有投机取巧、敷衍了事的心态。
例如,2017年韩美制药公司肺癌药物奥姆替尼(olmutinib)因延迟上报临床试验期间发生的患者致命的不良反应,涉嫌违反了临床试验安全监测的法规,最后被禁止上市销售。
因此,药企需要杜绝“糊弄过关”的侥幸心理,秉持严谨的研发态度,在临床试验阶段扎扎实实按照方案和GCP的要求,收集全面可靠的药品有效性、安全性数据,才能最终造福患者、赢得患者,从而赢得市场,行稳致远。
此内容为《医药经济报》融媒体平台原创。未经《医药经济报》授权,不得以任何方式加以使用, 包括转载、摘编、复制或建立镜像。如需获得授权请事前主动联系:020-37886610或020-37886753;yyjjb@21cn.com。





